
Research
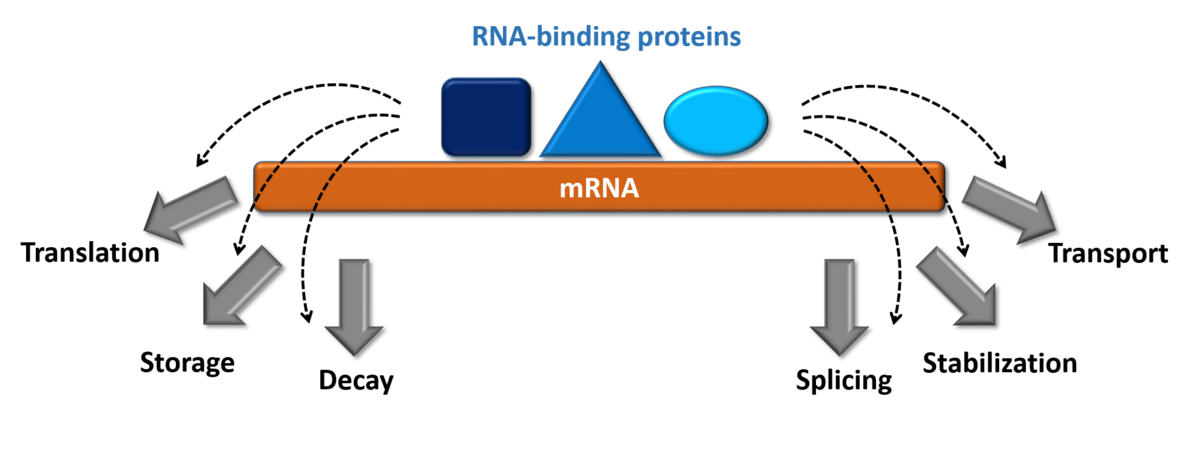
How do proteins achieve the necessary specificity in RNA-recognition during gene regulation?
Content to come...

Specific RNA fold recognition. Content to come...
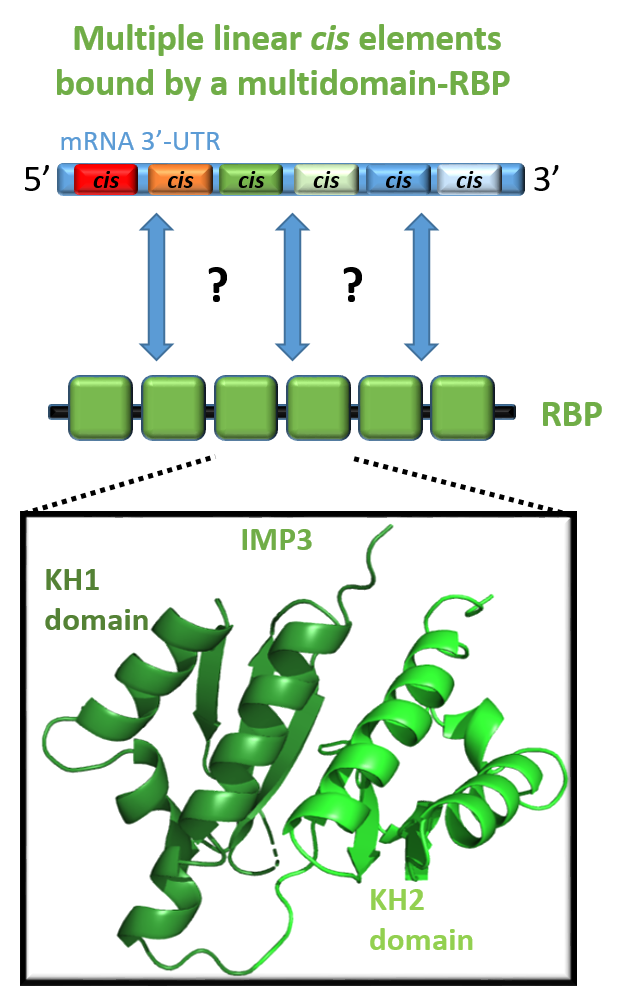
The specific recognition of linear RNAs by multi-domain RNA-binding proteins. Content to come...
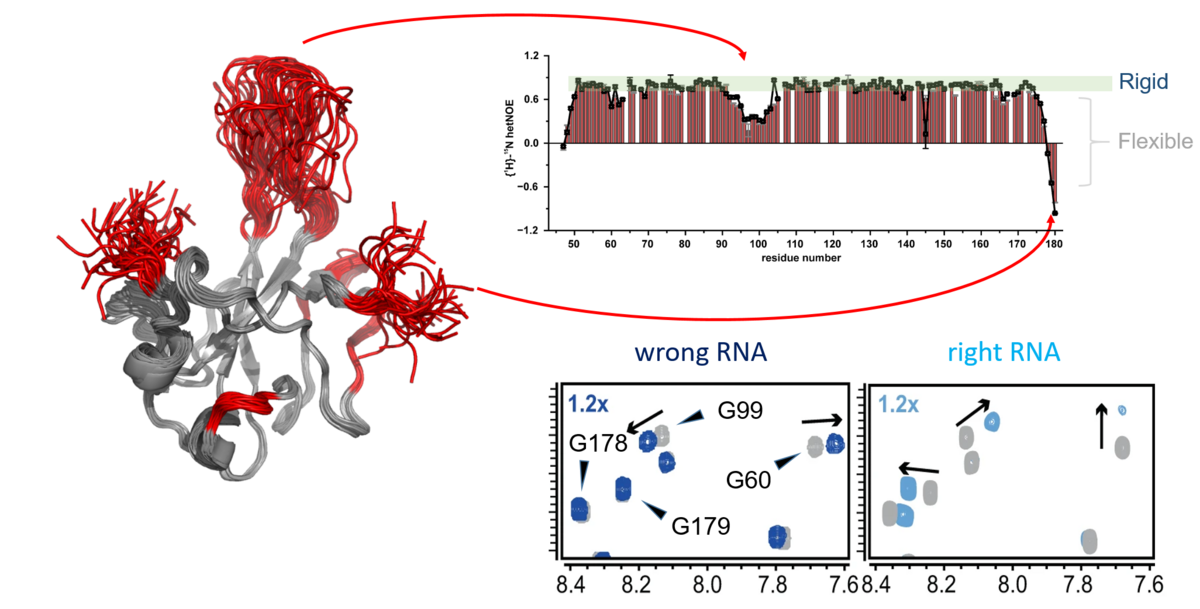
Virus-virus and virus-host RNPs. Content to come...
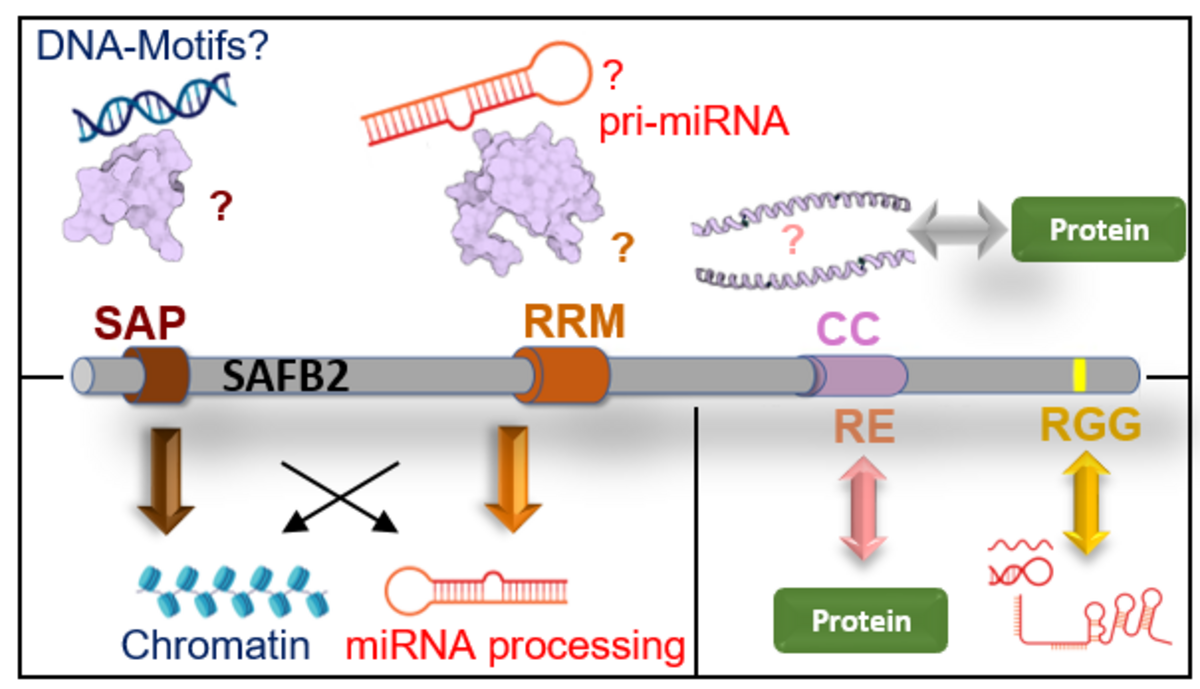
Dual nucleic acid-binding proteins: DRBPs. Content to come...

Structures of regulatory RNA hubs in their full native contexts. Content to come...